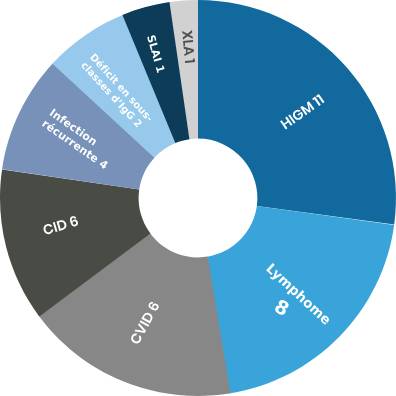
Lorsque les principaux symptômes sont des cytopénies auto-immunes, une lymphadénopathie, une splénomégalie, une hépatomégalie, une hyperplasie lymphoïde nodulaire ou un lymphome, le diagnostic peut être un trouble hématologique tel que le syndrome lymphoprolifératif avec auto-immunité (SLAI) et le syndrome d’Evans.